
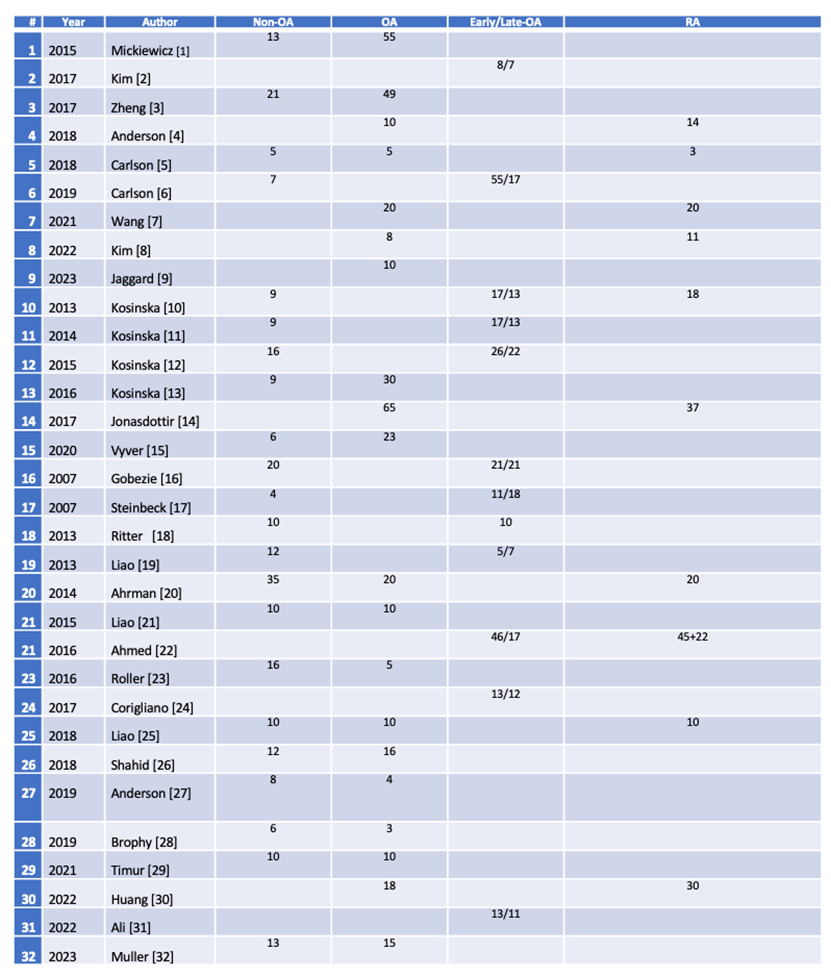
Multi-omics integrates genomic, transcriptomic, proteomic, metabolomic, lipidomic and epigenomic outcomes using sophisticated instruments and AI based software of biological systems [1]. Unlike traditional single-omics, multi-omics enables the investigation of complex interactions and dynamic relationships between different molecular layers in normal and pathological conditions. It analyzes complex data to find novel associations between biological entities, recognizes relevant biomarkers and builds detailed marker sets of disease and conditions. The OA consortium (https://www.genetics-osteoarthritis.com/themenmenue/home/index.html) focuses on genetic footprints of the disease. The foundation for the national institute of health biomarker consortium, osteoarthritis biomarkers project (https://fnih.org/our-programs/biomarkers-consortium-osteoarthritis-biomarkers-project/) defined several wet biomarkers that can be used for the diagnosis and progression of OA [2]. Eight serum protein biomarkers were identified to differentiate knee OA patients from normal individuals [3]. Six serum peptides corresponding to six proteins could predict the development and/or progression of OA [4]. Several adipokines such as leptin, adiponectin and visfatin were proposed as OA biomarkers stressing the importance of correlating comorbidities in patients [5]. Skeletal muscle released myokines and their correlation with degree of physical activity are also considered as biomarkers for the onset and treatment follow up of OA [6]. These studies however were limited mostly to the serum but not the SJF. Metabolome and lipidome studies were also carried out in limited patient groups. A recent literature search highlighted the need for such studies for SJF (Table).
Table. Multi-Omics Studies from the Synovial Joint Fluid in Limited Group of Osteoarthritis Patients.
scRNA-seq offers unprecedented insights into the cellular and molecular landscape to knee OA research. It provides a high-resolution view of cellular heterogeneity and disease progression by studying the diverse cell types. Studying these cell types could additionally help the identification of new therapeutic targets and design personalized treatment strategies. A study [7] identified seven distinct chondrocyte populations with unique gene expression profiles in human OA cartilage, including previously uncharacterized phenotypes, highlighting the functional diversity of these cells during disease progression. That study also identified a novel inflammatory/proliferative cluster, characterized by cells with a high expression of pro-inflammatory genes, highlighting the intricate interplay between inflammation and chondrocyte dysfunction in OA. They finally discovered key biomarkers and gene expression patterns associated with chondrocyte differentiation trajectories, suggesting potential therapeutic targets. Another study [8] identified three distinct chondrocyte subpopulations, namely, hypertrophic chondrocytes (HTCs), homeostatic chondrocytes (HomCs) and fibrocartilage chondrocytes (FCs). They showed that unique gene expression profiles can be used to characterize each subpopulation, suggesting specialized roles in OA pathogenesis. They also determined key biomarkers and gene expression patterns associated with chondrocyte differentiation trajectories, facilitating early diagnosis and enabling potential intervention points to modulate disease progression and monitor the treatment efficacy. One recent study [9] focused on effector chondrocytes (ECs) and FCs to better evaluate the disease severity because these subtypes are associated with higher disease severity and more robust immunogenicity. The authors showed that ECs play a central role in activity in immune-related pathways and are involved in immunoregulation through interactions with other cell types via signaling pathways. FCs on the other hand show higher metabolic activity, playing a central role in fibrillogenesis and cartilage tissue repair through the EGF and VEGF signaling pathways. That study also identified canonical gene markers for ECs (CHRDL2, DSC2) and FCs (COL1A1, COL14A1, IFI27, THY1) that could predict OA severity. Another study [10] focused on the cellular heterogeneity within the synovial tissues of patients with OA, identifying five distinct immune cell subpopulations: macrophages, dendritic cells, T cells, monocytes and B cells. Notably, they unveiled the main evolution path in the inflammatory microenvironment of the synovial tissue through pseudo-time analysis. They also explored intercellular communication and identified key differentially expressed genes and pathways associated with the cell subpopulations. The study also validated the role of two genes, NR4A1 and NR4A2, in OA progression using in vitro experiments. One study [11] investigated the dynamic changes in the immune cell landscape within the knee joint following an anterior cruciate ligament injury, profiling immune cells at various time points after the injury. The most dramatic changes after injury were observed in different monocyte and macrophage subpopulations, which significantly expanded. That study also characterized injury-induced changes in neutrophils and lymphocytes. Neutrophils in particular seemed to be a significant source of inflammatory cytokines and matrix-degrading enzymes. Li et al. investigated the molecular mechanisms underlying hand OA using a combination of scRNA-seq and population-based studies [12]. They identified 13 chondrocyte subpopulations, including a subpopulation of inflammatory chondrocytes characterized by the expression of inflammatory response and immune system-related genes, confirming the results obtained by Ji et al. Notably, they conducted a Mendelian randomization study using UK Biobank data and a cross-sectional study using data from the Xiangya Osteoarthritis Study to validate their findings. The results showed that genetic predisposition to higher FTH1 expression increased the risk of hand OA and high serum ferritin levels encoded by FTH1 were associated with a higher prevalence of hand OA. Chen et al. analysed gene expression profiles from four datasets to identify differentially expressed genes (DEGs) between OA and control groups [13]. They performed immune cell infiltration analysis, weighted gene co-expression network analysis and other analyses using different machine learning algorithms to identify main immune-related DEGs and construct a diagnostic model. The top 12 immune-related DEGs were used to construct a nomogram diagnostic model able to predict the OA disease stage. Chen et al. [14] employed an integrative approach, combining scRNA-seq and bulk RNA sequencing (RNA-seq) data, to identify key pyroptosis-related genes and pathways involved in OA pathogenesis. The authors identified hub genes associated with pyroptosis, a form of programmed cell death associated with inflammation, including CASP6, NOD1, and PYCARD, which were also found to be independent prognostic factors for OA. These hub genes were enriched in notch signaling and oxidative phosphorylation pathways, suggesting their potential as therapeutic targets. Fan et al. [15] proposed a comprehensive investigation of the cellular heterogeneity and spatial organization of chondrocytes in human knee articular cartilage. This study employed a multi-omics approach, integrating scRNA-seq and spatially resolved transcriptomic data to provide a high-resolution view of the cartilage landscape in both OA and non-OA conditions. The authors showed that most OA-associated differentially expressed genes reside in the cartilage’s articular surface and superficial zone. Among the different chondrocyte populations, the authors found that pre hypertrophic chondrocytes (preHTCs) and hypertrophic chondrocytes (HTCs) are critical OA cell populations, where preHTCs show transcriptional associations with OA, while HTCs are genetically linked to OA risk variants.
Focusing on the immunogenicity of chondrocyte subpopulations allows us to understand the immune responses in OA pathogenesis better. This approach opens new possibilities for immunomodulatory interventions, as well as for facilitating early diagnosis and monitoring of treatment response. Identifying prognostic biomarkers and potential therapeutic targets can pave the way for developing promising diagnostic and treatment strategies for OA.
References
[1] Vilanova C, Porcar M. Are multi-omics enough? Nature Microbiology. 2016;1:16101. doi:10.1038/nmicrobiol.2016.101
[2] Kraus VB, Hargrove DE, Hunter DJ, Renner JB, Jordan JM. Establishment of reference intervals for osteoarthritis-related soluble biomarkers: the FNIH/OARSI OA biomarker consortium. Ann Rheum Dis 2017;76:179-185.
[3] Kraus VB, Reed A, Soderblom EJ, Golightly YM, Nelson AE, Li YJ. Serum proteomic biomarkers diagnostic of knee osteoarthritis. Osteoarthritis and Cartilage 2024;32:329-337.
[4] Kraus VB, Sun S, Reed A, Soderblom EJ, Moseley MA, Zhou K, Jain V, Arden N, Li YJ. An osteoarthritis pathophysiological continuum revealed by molecular biomarkers. Sci Adv 2024;10:eadj6814.
[5] Conde, J., Scotece, M., Gómez, R., et al. (2011). Adipokines and osteoarthritis: novel molecules involved in the pathogenesis and progression of disease. Arthritis Research & Therapy, 13(4), 237
[6] Ning K, Wang Z, Zhang XA. Exercise-induced modulation of myokine irisin in bone and cartilage tissue-Positive effects on osteoarthritis: A narrative review. Front Aging Neurosci. 2022 Aug 19;14:934406.
[7] Ji, Q., Zheng, Y., Zhang, G., Hu, Y., Fan, X., Hou, Y., … & Tang, F. (2019). Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Annals of the rheumatic diseases, 78(1), 100-110.
[8] Zhang, X., Huang, N., Huang, R., Wang, L., Ke, Q., Cai, L., & Wu, S. (2020). Single-cell rna seq analysis identifies the biomarkers and differentiation of chondrocyte in human osteoarthritis. American Journal of Translational Research, 12(11), 7326.
[9] Hu, X., Li, Z., Ji, M., Lin, Y., Chen, Y., & Lu, J. (2022). Identification of cellular heterogeneity and immunogenicity of chondrocytes via single-cell RNA sequencing technique in human osteoarthritis. Frontiers in Pharmacology, 13, 1004766.
[10] Liu, W., Chen, Y., Zeng, G., Yang, S., Yang, T., Ma, M., & Song, W. (2022). Single-cell profiles of age-related osteoarthritis uncover underlying heterogeneity associated with disease progression. Frontiers in Molecular Biosciences, 8, 748360.
[11] Sebastian, A., Hum, N. R., McCool, J. L., Wilson, S. P., Murugesh, D. K., Martin, K. A., … & Loots, G. G. (2022). Single-cell RNA-Seq reveals changes in immune landscape in post-traumatic osteoarthritis. Frontiers in immunology, 13, 938075.
[12] Li, H., Jiang, X., Xiao, Y., Zhang, Y., Zhang, W., Doherty, M., … & Lei, G. (2023). Combining single-cell RNA sequencing and population-based studies reveals hand osteoarthritis-associated chondrocyte subpopulations and pathways. Bone Research, 11(1), 58.
[13] Chen, B., Lin, C., Jin, X., Zhang, X., Yang, K., Wang, J., … & Meng, Z. (2024). Construction of a diagnostic model for osteoarthritis based on transcriptomic immune-related genes. Heliyon, 10(1).
[14] Chen, Y., Zhang, Y., Ge, Y., & Ren, H. (2023). Integrated single-cell and bulk RNA sequencing analysis identified pyroptosis-related signature for diagnosis and prognosis in osteoarthritis. Scientific Reports, 13(1), 17757.
[15] Fan, Y., Bian, X., Meng, X., Li, L., Fu, L., Zhang, Y., … & Sun, S. (2024). Unveiling inflammatory and prehypertrophic cell populations as key contributors to knee cartilage degeneration in osteoarthritis using multi-omics data integration. Annals of the Rheumatic Diseases, 83(7), 926-944.